

La dinamica molecolare (MD) è un metodo di simulazione numerico utile ad analizzare i moti atomici e molecolari. Gli atomi e le molecole possono interagire tra loro per un periodo di tempo più o meno limitato. La somma delle varie interazioni tra molecole permette di descrivere la cosiddetta “evoluzione” dinamica del sistema. Nella versione più comune, le traiettorie degli atomi e delle molecole sono determinate risolvendo numericamente le equazioni Newtoniane del moto per ognuna delle particelle che compongono il sistema. Le forze tra le particelle e le loro energie potenziali sono spesso calcolate usando i cosiddetti “potenziali interatomici” o “campi di forza meccanici molecolari”. La possibilità di conoscere l’evoluzione dinamica del sistema può essere utile in molti ambiti industriali, dalla fisica, alla chimica, dalla scienza dei materiali alla biofisica.
Poiché i sistemi molecolari (di fatto un qualsiasi sistema fisico su scala nanoscopica) si compongono sempre di un numero molto elevato di particelle, è impossibile determinare analiticamente le proprietà di tali sistemi. La simulazione MD aggira questo problema tecnico mediante utilizzo di metodi numerici.
Le simulazioni di dinamica molecolare
Per i sistemi che obbediscono all’ipotesi ergodica (dopo un tempo sufficientemente lungo, il tempo speso da una particella in un volume nello spazio delle fasi di microstati della stessa energia è proporzionale al volume stesso; equivalentemente alle condizioni termodinamiche, il suo stato può essere uno qualunque di quelli che soddisfano le condizioni macroscopiche del sistema), l’evoluzione di una simulazione di dinamica molecolare può essere utilizzata per determinare le proprietà termodinamiche macroscopiche del sistema: le medie temporali di un sistema ergodico corrispondono alle medie di ensemble dei sistemi micro-canonici. La dinamica molecolare è talvolta anche definita “meccanica statistica” o “visione di Laplace della meccanica Newtoniana” e permette dunque la comprensione del moto molecolare su scala atomica.
La storia della dinamica molecolare
La MD è stata originariamente sviluppata nei primi anni ‘50, in seguito agli sviluppi delle simulazioni Monte Carlo, che a loro volta risalgono al XVIII secolo. L’interesse per l’evoluzione temporale dei sistemi a N-corpi risale però a molto prima, al Diciassettesimo secolo, a partire dai tempi di Newton. Tale interesse è continuato per tutto il secolo successivo con focus principale legato alla meccanica celeste ed alla stabilità del sistema solare. Molti dei metodi numerici utilizzati oggigiorno sono stati sviluppati durante quel periodo, quindi molti anni prima dell’invenzione del calcolatore. A titolo di esempio l’algoritmo di integrazione più comunemente utilizzato oggi (i.e. l’algoritmo di integrazione Verlet) fu concepito già nel 1791 da Jean Baptiste Joseph Delambre. I calcoli con questi algoritmi possono essere considerati una prima versione manuale della odierna MD.
A partire dal 1941, l’integrazione delle equazioni multi-corpo è stata possibile sfruttando i primi computer analogici. Alcuni ricercatori intrapresero l’arduo compito di modellare il moto atomico con modelli fisici basati su sfere macroscopiche. Lo scopo era quello di disporre tali corpi rigidi così da replicare la struttura di un liquido e utilizzarla per esaminarne il comportamento. J.D. Bernal, nel 1962 disse: “… Ho preso un certo numero di palline di gomma e le ho incollate insieme con aste di una selezione di lunghezze diverse che vanno da 2,75 a 4 pollici. Ho cercato di farlo in primo luogo nel modo più casuale possibile, lavorando nel mio ufficio, venendo interrotto ogni cinque minuti circa e non ricordando cosa avessi fatto prima dell’interruzione.”
In seguito alla scoperta di particelle microscopiche e allo sviluppo dei computer, l’interesse per la MD si espanse oltre l’ambito dei sistemi gravitazionali aprendosi allo studio delle proprietà della materia. In tal senso, una delle prime simulazioni di un sistema a N-corpi è stata effettuata da Enrico Fermi per comprendere le origini dell’irreversibilità in natura. Nel 1957, Alder e Wainwright usarono un computer (IBM 704) per simulare le collisioni perfettamente elastiche tra sfere dure. Con il 1960 si arriva a quella che è considerata la prima simulazione realistica della materia, a cura di Gibson et al.. Essi simularono il danno causato da radiazioni del rame solido usando un tipo di interazione repulsiva di Born-Mayer combinata ad una forza superficiale coesa.
Nel 1964, Rahman pubblicò il risultato di simulazioni di argon liquido che utilizzavano un potenziale di Lennard-Jones; le stime delle proprietà del sistema, come ad esempio il coefficiente di auto-diffusione, per la prima volta risultavano allineati ai dati sperimentali. Oggi, il potenziale di Lennard-Jones è ancora uno dei potenziali intermolecolari più frequentemente usati.
Un po’ di teoria
La progettazione di una simulazione di dinamica molecolare deve tenere conto della potenza di calcolo disponibile. Le dimensioni della simulazione (numero di particelle), il passo temporale e la durata totale del tempo devono essere scelti in modo che il calcolo possa terminare entro un periodo di tempo ragionevole. Tuttavia, le simulazioni devono essere sufficientemente lunghe da essere pertinenti alle scale temporali dei processi naturali studiati.
Per trarre conclusioni statisticamente valide dalle simulazioni, l’arco di tempo simulato deve corrispondere alla cinetica del processo naturale. Altrimenti sarebbe come cercare trarre conclusioni su come un individuo cammini osservandone solo un passo. Purtroppo, anche con hardware performanti, simulare qualche nano-secondo può richiedere giorni o mesi di tempo calcolo.
Gli algoritmi di parallelizzazione consentono di distribuire il carico tra le CPU. Durante una simulazione MD classica, il compito più impegnativo per la CPU è la valutazione del potenziale in funzione delle coordinate interne delle particelle. All’interno di questa valutazione energetica, la parte più onerosa è quella legata al calcolo delle interazioni elettrostatiche e di van der Waals. L’onere computazionale può essere ridotto impiegando metodi elettrostatici come la sommatoria di Ewald (una sorta di media spaziale).
Un altro fattore che influisce sul tempo di calcolo necessario per una simulazione è la dimensione del passo temporale di integrazione. Si tratta del tempo che intercorre tra due valutazioni successive del potenziale interatomico. Il passo temporale deve essere scelto abbastanza piccolo da evitare errori di discretizzazione (cioè, più piccolo del periodo relativo alla frequenza vibrazionale più elevata).
I tempi tipici per le simulazioni MD classiche sono dell’ordine di 1 femtosecondo (10-15 s). Questo valore può essere esteso utilizzando algoritmi che approssimano le vibrazioni degli atomi più rapidi (ad esempio, gli ioni idrogeno). Sono stati sviluppati anche metodi a scala temporale multipla, che consentono tempi più lunghi tra gli aggiornamenti delle forze a lungo raggio (tra atomi distanti tra loro). Inoltre, la dimensione del dominio di simulazione deve essere sufficientemente grande per evitare errori dovuti alle condizioni al contorno. Le condizioni al contorno sono spesso trattate scegliendo valori fissi oppure trattate come periodiche.
Gli ensemble:
L’intero universo è governato dalle leggi della termodinamica attraverso il trasferimento di energia. Questo processo è troppo complesso per essere considerato direttamente nelle simulazioni MD; si considerano invece diverse semplificazioni in cui una parte dell’universo (cioè il sistema in esame) viene considerata isolata dal resto dell’ambiente circostante. Il sistema ospita il processo o lo stato termodinamico in esame, mentre l’ambiente circostante contiene tutto il resto.
Il sistema può essere descritto utilizzando un insieme (ensemble), noto anche come insieme statistico, che è un’idealizzazione costituita da un gran numero di stati di un sistema in cui potrebbe trovarsi il sistema reale, considerati tutti contemporaneamente. Un insieme termodinamico è un insieme statistico in equilibrio. Fornisce un modo per derivare le proprietà di un sistema termodinamico reale dalle leggi della meccanica classica e quantistica.
È possibile considerare diversi sistemi con diversi gradi di separazione dall’ambiente circostante, da completamente isolati a completamente aperti all’ambiente circostante. Tuttavia, ai fini delle simulazioni molecolari in ambito meccanico, l’insieme più comunemente utilizzato è il cosiddetto ensemble NVE (o micro-canonico). Nell’ensemble micro-canonico, il sistema è isolato dalle variazioni di moli (N), volume (V) ed energia (E). Corrisponde a un processo adiabatico senza scambio di calore. Una traiettoria di dinamica molecolare micro-canonica può essere vista come uno scambio di energia potenziale e cinetica, con l’energia totale conservata.
Campi di applicazione e limiti
Utilizzato per la prima volta in fisica teorica, il metodo MD ha guadagnato popolarità in molti altri ambiti. La dinamica molecolare è frequentemente impiegata per raffinare strutture tridimensionali di proteine e altre macromolecole sulla base di vincoli sperimentali dalla cristallografia a raggi X o dalla spettroscopia NMR. In fisica, la MD è usata per esaminare la dinamica di fenomeni a livello atomico che non possono essere osservati direttamente, come la crescita di film sottili, o per esaminare le proprietà fisiche di dispositivi nanotecnologici che con le tecnologie attuali non possono ancora essere realizzati. In biofisica e biologia strutturale, il metodo è frequentemente applicato per studiare i moti delle macromolecole quali proteine e acidi nucleici, che possono essere utili per interpretare i risultati di alcuni esperimenti biofisici e per modellare le interazioni con altre molecole. In linea di principio la MD può essere utilizzata per la predizione ab initio della struttura proteica simulando il ripiegamento della catena polipeptidica da bobina casuale.
Più recentemente, grazie alla crescente potenza di calcolo disponibile, la dinamica molecolare sta trovando sempre più spazio anche a livello meccanico. L’approccio molecolare, infatti, permette di studiare in modo accurato fenomeni fisici di primario interesse per la progettazione meccanica industriale, che spaziano dalla tribologia alla propagazione di cricche per fatica.

La dinamica molecolare per lo studio delle propagazioni di cricca nei materiali metallici
La fatica e la propagazione di cricche che possono portare al cedimento dei componenti meccanici in esercizio sono tematiche centrali nel processo di progettazione ingegneristico. Se storicamente il problema della fatica è dapprima stato affrontato su scala macroscopica mediante approcci classici (quali diagramma di Wöhler) e successivamente su scala microscopica mediante la cosiddetta meccanica della frattura. Oggigiorno la frontiera della ricerca sulla fatica si può spingere a livello nanoscopico proprio grazie alla dinamica molecolare.
È noto come l’accrescimento granulare e la conseguente microstruttura giochi un ruolo importante in termini di proprietà meccaniche dei materiali metallici. Le simulazioni di dinamica molecolare, ad esempio, permettono di studiare i meccanismi di propagazione delle cricche intergranulari lungo i bordi-grano in qualsiasi condizione di carico e forniscono le modalità di deformazione e la distribuzione delle sollecitazioni in prossimità dei difetti su scala atomica durante tutto il ciclo di carico imposto. Esempi di simulazioni MD di propagazione di cricche intergranulari sono state eseguite in molti metalli con struttura cubica a corpo centrato (BCC) e cubica a facce centrate (FCC), come Fe, Cu, Al e Ni. Dai risultati di questi studi è emerso come il meccanismo di propagazione delle cricche sia influenzato dalla struttura GB (angolo di disorientamento), dalla direzione di propagazione, dal livello di segregazione e dalla temperatura.
Le simulazioni MD sono anche state frequentemente applicate per studiare la propagazione delle cricche nei cristalli di Mg. Le simulazioni della crescita di cricche in condizioni di carico di trazione monotono e ciclico in cristalli singoli di Mg hanno mostrato come la deformazione plastica attorno all’apice della cricca dipenda dal piano cristallografico e dalla temperatura.
La dinamica molecolare per lo studio dell’usura adesiva
Un altro ambito di applicazione della MD è il campo della tribologia. In particolare, per lo studio dell’usura adesiva. Questa si manifesta in conseguenza di una saldatura a freddo tra asperità superficiali durante il contatto con strisciamento. Studi recenti dimostrano come esista una dimensione critica delle asperità (rugosità), al di sopra della quale tali asperità superficiali si staccano dalla superficie per fessurazione sub-superficiale.
I modelli continui non sono in grado di simulare esplicitamente il processo di usura a causa della complessità data dalle grandi deformazioni, della nucleazione di cricche e rimozione del materiale. L’alternativa può essere rappresentata da una modellazione atomistica discreta, approccio in grado di gestire queste caratteristiche, eventualmente combinata, per ridurre l’onere computazionale, ad un approccio continuo (FEM) [1].
Si considerino due piastre tridimensionali con due asperità cilindriche di dimensioni 3000 × 1000 × 150 Angstrom estese nella direzione del cristallo. Le asperità con raggio di 200 Å e le aree annesse (piastre di 3000 × 250 × 150 Angstrom) sono modellate con approccio atomistico. Per la modellazione di ciascuna piastra sono stati utilizzati 135 elementi e 320 nodi. Viene imposto uno spostamento di 0,08 Å in direzione x alla sola piastra superiore.
Il passo temporale è 0,003 s e la simulazione è stata eseguita per 300000 fs.
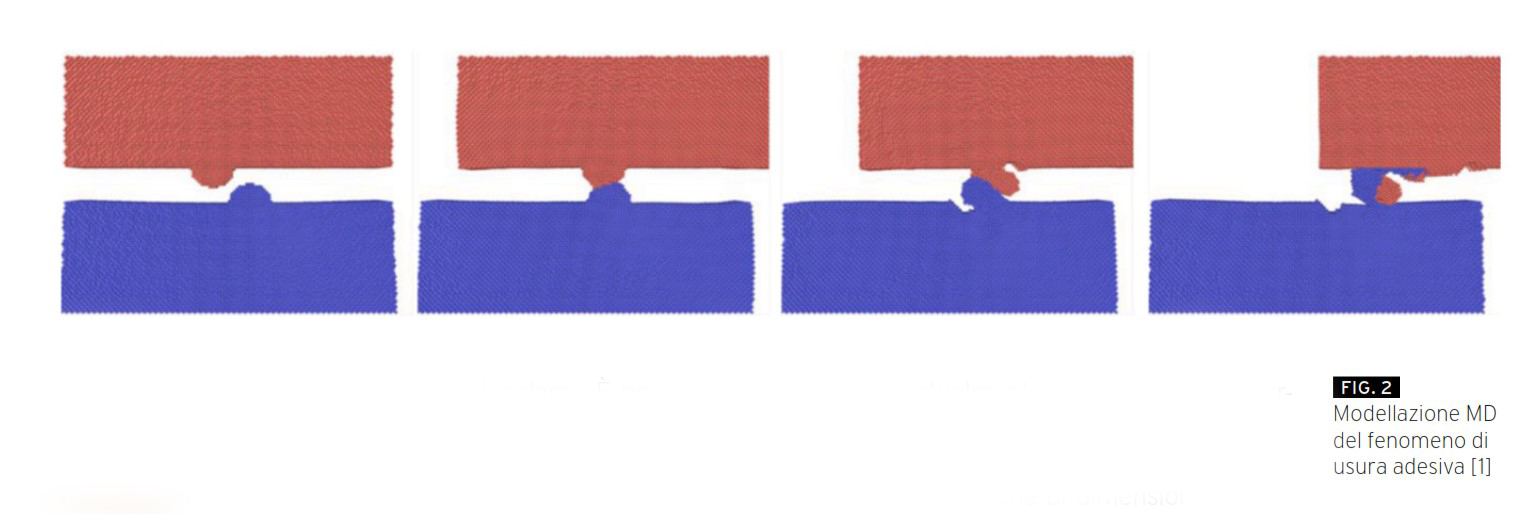
La Figura 2 mostra il meccanismo di formazione dei detriti dopo la collisione tra due asperità superficiali. Il tempo di simulazione per il modello atomistico è stato pari a 1680 min. Da tale simulazione è anche stato possibile prevedere la forza tangenziale (forza di attrito). A livello di danneggiamento, si osserva come le cricche sub-superficiali nucleino e si propaghinono al di sotto delle asperità. Con questo approccio è dunque possibile anche meglio comprendere i fenomeni fisici alla base del cedimento ed eventualmente studiare l’impatto di azioni correttive (riduzione Ra, applicazione di un trattamento superficiale etc.) sulla durata e sull’attrito tra corpi a contatto.
Conclusioni
Le simulazioni MD hanno un vastissimo campo di applicazione e permettono i studiare in modo molto dettagliato il comportamento a livello molecolare/atomico. In ambito meccanico/ingegneristico, applicazioni di particolare interesse sono rappresentate dalla frattura e dalla tribologia. A titolo esemplificativo stati riportati due diversi esempi, una frattura ed una usura adesiva. L’intento era quello di dimostrare l’applicabilità e l’efficienza del metodo proposto.
Bibliografia
[1] Niknafs, S., Silani, M., Concli, F., & Aghababaei, R. (2022). A coarse-grained concurrent multiscale method for simulating brittle fracture. International Journal of Solids and Structures, 254, 111898.






